 = K/PTot2.
(5)
= K/PTot2.
(5)Membrane reactors achieve efficiencies by combining in one unit a reactor that generates a product with a semipermeable membrane that extracts it. The result is a more compact design plus greater conversion. Removal of a product increases the residence time for a given volume of reactor and drives equilibrium-limited reactions towards completion. This latter advantage is reviewed excellently by Armor1,2 while the former has been, largely, ignored. In my opinion, a yet-larger advantage of membrane reactors is that they expand the allowed range of temperatures and pressures for a reaction. Membrane reactors fundamentally change the pressure dependence of conversion in gas phase decomposition reactions so that the reactions are preferentially performed at high pressures rather than low. Higher pressures allow much smaller reactors and more efficient purification. Membrane reactors can be advantageous also for sequential endothermic and exothermic reactions, by using the product extraction to promote heat transfer. Enhanced heat transfer permits plug flow where CSTR designs would have been necessary otherwise. The net result is smaller reactors, lower capital costs, and often fewer side-reactions. These general benefits will be illustrated for methanol reforming to hydrogen.
An example application, methanol reforming:
Consider the methanol reforming reaction which is being pursued as a source of hydrogen to fuel fuel cells, e.g. for mobile electricity generation on boats, for military hospitals, and electric cars.

Fundamental Improvement in Pressure Dependence:
Performing the same reaction in a membrane reactor give much purer output
hydrogen and produces a fundamental improvement in the way that extent
of reaction is affected by pressure. Instead of benefiting from low pressures,
this reaction, when performed in a membrane reactor benefit from being
performed at the highest pressures available, as will be shown below.
The equilibrium constant for the overall methanol reforming reaction
(1) is expressed in terms of partial pressure and mol fraction respectively
as
= K/PTot2.
(5)Clearly, the higher the total pressure, the lower the extent of reaction
at equilibrium.
In a membrane reactor, the hydrogen partial pressure becomes less of
a function of the extent of reaction, and more of a function of the backpressure
of the hydrogen outlet. (For mobile use, ß will about 1 atmosphere).
For the ideal membrane reactor, where transport resistance is minimal,
the hydrogen partial pressure equals this hydrogen backpressure. We can
define a constant ß as this hydrogen back-pressure and derive
extent equations for the ideal membrane reactor. Substituting ß for
the hydrogen partial pressure, equation 4 becomes:
K = ß3 [yCO2] / [yH2O][yCH3OH]PTot
(6)
As before, we will consider the extent of reaction for an feed of one
mol each of methanol and water, and will define the extent of the reaction
as equaling the mol conversion to CO2. For this case, the number of moles
of hydrogen in the reactor is ß(2-extent)/(PTot -ß),
and the total number of moles is {2-extent +ß(2-extent)/(PTot
-ß)}. At equilibrium, Eqn. 6 becomes:
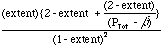 = PTot K/ß3.
(7)
= PTot K/ß3.
(7)Equation 7 provides two insights into the advantages of membrane reactors: (1) lowering the product pressure, ß strongly increases the reactor conversion, and (2) for the membrane reactor the higher the total pressure, the greater the conversion. This pressure dependence is fundamentally different from that observed in the normal reactor, Eqn. 5, and allows tremendous design improvements. High total pressure not only drives conversion, but also decreases the volumetric flow rate for gas-phase reactions (volumetric flow rate for gases decreases inversely with overall pressure at a given molar flow rate). A decreased volumetric flow rate increases the residence time, or allows a much smaller reactor for a given residence time. To get these advantages with a membrane reactor, one needs a good catalyst and sufficient membrane to rough equality between the back-pressure, ß and the product partial pressure. An appropriate reactor design also must be chosen to allow sufficient heat transfer and energy management.
Membrane Reactor Designs:
Two membrane reactors for use with methanol reforming are shown in Figures
2 and 3; the one in Figure 2 has already been built and tested, the one
in Figure 3 was proposed but never built. In both designs, a methanol water
mixture enters the outer annulus, flows up while being heated, and flows
downward inside the reactor core. The reactor in Figure 2 used 100 cc of
catalyst. Heat was provided by electrical heating tape augmented by heat-exchange
between the core and the annulus. Product hydrogen was purified and removed
through seven 5.5î long REB Research membrane tubes. The reactor ran continuousely
for one month on a feed on methanol-water mixture with a hydrogen output
of 800 cc/min at 150 psi and 265 °C. The relatively high
operating pressure meant that the reactor could be much smaller than an
equivalent sequential reactor-separator. Further, the output purity from
the membranes was 100% so that no subsequent hydrogen purification was
needed. The weight and volume advantages could be significant for vehicular
use, providing more passenger space, a quicker start-up, and better acceleration.
There is a downside to this design and to membrane reactors in general.
Driving hydrogen (or any product) through a membrane requires a partial-pressure
driving force and a partial pressure of gas in the reactor vent at least
as high as that of the pure collected gas. In the ideal case above, ß(2-extent)/(PTot
-ß) moles of hydrogen had to be vented with the waste gas for every
two mols of feed. When the actual unit was tested at Tufts University, 80% of the theoretical, stoichiometric
hydrogen was recovered, and 20% was lost. This recovery is higher than with normal sequential reformer-membrane separators partially because
of the inherent advantages of membrane reactors, and partially because the membrane was good enough to
keep a low hydrogen partial pressure within the reactor. Still,
even with a perfect catalyst and membrane at least 5% of the potential
hydrogen would be lost.
Another deficiency of the reactor design in Figure 2 is that it was
heated electrically. For commercial use, we will not be able to afford
electric heating. Further, we will want to be more fuel efficient and cleaner.
One improvement in this direction is to combust the waste gas, including
the waste hydrogen, in a separate reactor to provide heat to run the reformer.
A membrane reactor that does this is shown in schematic in Figure 3, patent
pending. It is really just the design in Figure 2 with an attached catalytic
combustor and a flow restrictor to prevent back-burning. Methanol plus
water enters at the bottom of the outer annulus and is vaporized by heat
recovered from cooling the product hydrogen and the shift reaction. The
vapor goes from the annulus to the upper section of the reformer through
the portal shown at the upper right. It is further heated in the reformer
by raffinate gas combustion. The unit shows an electrical heater as well,
but this would be used only for start-up. 
As with the design in Figure 2, the first few inches inside the reactor
in Figure 3 is a decomposition zone, where methanol is converted to CO
and H2 via reaction 2. This is followed by the membrane-reactor zone where
the water-gas shift reaction takes place. As before, removing hydrogen
helps drive the reaction at high pressure by keeping the hydrogen partial
pressure below that of carbon monoxide and water. The result is a smaller
lighter unit than would be possible otherwise. Burning the waste gas improves
the overall efficiency while removing any remnant CO. With the appropriate
membranes, discussed below, this unit can easily provide purer hydrogen
than is available with partial oxidation.
Other Reactions:
Several researcher have demonstrated size and operation advantages of membrane reactors for a variety of reactions beyond the example case of methanol reforming. A group at headed by Barry Pruden at the University of Calgary has gotten good results in a fluidized bed steam-methane reformer using REB membranes at 600 -700°C5. Dr. Pruden thinks the very high purity hydrogen produced by this membrane reformer may be cheaper than low purity hydrogen produced by current sequential reformer - PSA units; he is constructing a pilot plant demonstration. Also, a group headed by T. Tsotsis at USC has demonstrated efficient steam reforming of ethane to ethylene at 700 °C instead of the typical 850 °C6. More daringly, Aspen Systems, co. of Marlborough, Mass., is currently testing a membrane reactor to convert diesel fuel and water to hydrogen at about 500°C. In all these cases, the main benefit from the membrane is not that it drives the reaction, but that it permits the use of a smaller reactor and separator, and that it allows operation at a greater residence time, at lower temperatures and higher pressures. The benefit is decreased reactor cost (cheaper materials, smaller vessel and less catalyst), fewer side reactions and improved heat transfer. Further capital savings may accrue from less expensive piping, fittings and siting. None of these studies are beyond the pilot plant stage though.
Membrane Choices:
The REB Research membranes used in most of the studies mentioned are
made by palladium coating high permeability alloy tubes. The manufacture
and principals are described elsewhere7 and the performance
is shown in figure 4. The general advantage of membrane reactors is independent
of the use of our membranes, though and Tsotsisís application was performed Figure 4.
Figure 4.
with a single porous ceramic tube6. This is not to say that
any membrane is appropriate for any reaction. Palladium silver membranes
are an established standard, with a great deal of experimental evidence
to show that they work. Further, fabrication technology for palladium silver
is very well developed so that units of 30,000 cm2 are available.
Our palladium-coated composite membranes are less developed and we have
made modules with only as much as 2000 cm2 of surface. Still
these units provide about 3 times more flux per unit area than palladium
silver at 400 °C, and show approximately three times the flux per dollar.
The flux advantage increases at lower temperatures and in the presence
of CO so that for many applications, they allow a significantly lower ß,
or optionally the use of significantly less surface area. Ceramic membranes
(coated and uncoated) show much higher flux than non-porous membranes when
operated at their ideal temperatures and without fouling ingredients. The
technology for sealing many ceramic membranes into a module for high temperature
operation is currently less developed than for the non-porous metallic
membranes.
Temperature and resistance to fouling are as critical as flux when
choosing a membrane. Palladium-coated metal membranes, and uncoated ceramics
have operated at temperatures between 100°C and 800 °C, although
a single substrate choice will not operate well at all these temperatures.
By contrast, palladium-silver and palladium-silver coated ceramics work
well in the narrower range between about 300°C and 425°C. Glass-coated
ceramic membranes work best between 600 and 700°C. Carbon-coated ceramics,
like those demonstrated by Rao and Sircar9 appear to work best
at room temperature and below.
Concerning fouling, palladium-silver does not foul with water but fouls
easily with even ppb levels of H2S. Ceramics, by contrast, are not affected
by H2S , but deteriorate in moist hot gas. Our Pd-coated metal membranes
are inbetween: they tolerate water below about 700°C, and up to 1 ppm
of H2S. Two newer membrane materials, palladium-copper and palladium-ruthenium,
are reported to be exceptionally resistant to water and sulfur, but the
flux is lower than palladium silverís.
Several promising membranes of palladium or palladium-silver on porous
ceramic or steel2 have been demonstrated. Flux and operating
pressure are as high as with palladium-coated refractory metals. Early
work produced small membranes of poor durability2, but this
is improving; the largest palladium-coated ceramic membranes now boast
nearly 300 cm2 in surface area and a selectivity intermediate
between ceramics and dense metals.
Acknowledgement: My profound thanks go to Dr. Ronald Mann of
the Royal Military Academy of Canada for helpful comments on this
manuscript.
References:
1. J.N. Armor, ěCatalysis with Permselective Inorganic
Membranesî J. Appl. Catalysis 49 (1989)1-25.
2. J.N. Armor, ěMembrane Catalysis: Where is it now, what needs to be done?î Catalysis Today 25 (1995) 199-207.
3. J.C.Amphlett, K.A.M.Creber, J.M.Davis, R.F.Mann,B.A.Peppley, and D.M.Stokes, ěHydrogen Production by Steam Reforming of Methanol for Polymer Electrolyte Fuel Cellsî, Int. J. Hydrogen Energy, 19 (1994) 131-137.
4. J.C.Amphlett, R.F.Mann, and B.A.Peppley, ěOn board Hydrogen Purification for Steam Reformation/PEM Fuel Cell Vehicle Power Plants,î Int. J. Hydrogen Energy, 21 (1996) 673-678.
5. S.Roy, M.T.Islam, B.B.Pruden, and R.E.Buxbaum, ěPalladium Coated, high Flux Tubular Membranes for Hydrogen Separation at high Temperatures and Differential Pressuresî 1997 SAE Fuels and Lubricants Meeting, Edmonton.
6. T.T.Tsotsis, A.M. Champagnie, S.P.Vasileiadis, Z.D. Ziaka, and R.G.Minet, ěThe Enhancement of Reaction Yield through The Use of High Temperature Membrane Reactorsî Sep. Sci. and Tech. 28 (1993) 397-422.
7. R.E.Buxbaum and A.B.Kinney, ěHydrogen Transport through Tubular Membranes of Palladium-Coated Tantalum and Niobiumî, I&EC Research 35(1996)530-537. More recent information available at http://www.rebresearch.com/products.htm.
8. J.C. Amphlett, R.F. Mann, and B.A. Peppley, ěPredicted Emissions From a Methanol-Fueled ěElectrochemical Automobile Engine Based on A PEM Fuel Cellî , Paper 952374, presented at the 1995 SAE International Fuels and Lubricants Meeting, Toronto Ont., October 1995.
9. M.B.Rao and S. Sircar, ěNanoporous Carbon Membranes
for Separation of Gas Mixtures by Selective Surface Flowî J. Membrane Sci.
85 (1993) 253-264.
THIS PAPER WAS PRESENTED AT THE 15TH BCC MEMBRANE PLANNING CONFERENCE, NEWTON MASS., OCT. 27-29, 1997, AND AT THE CANADIAN AICHE MEMBRANE SEPARATIONS MEETING, CALGARY ALBERTA, AUGUST, 1997.